




实验动物与比较医学 ›› 2024, Vol. 44 ›› Issue (2): 180-191.DOI: 10.12300/j.issn.1674-5817.2023.123
包方奇1( ), 屠海烨2, 方明笋3, 张倩3, 陈民利3(
), 屠海烨2, 方明笋3, 张倩3, 陈民利3( )()
)()
收稿日期:2023-09-01
修回日期:2023-11-24
出版日期:2024-04-25
发布日期:2024-04-25
通讯作者:
陈民利(1963—),女,硕士,博士研究生导师,教授,研究方向:实验动物与比较医学。E-mail: cmli991@126.com。ORCID: 0000-0003-1911-9397作者简介:包方奇(2000—),男,复旦大学临床五年制华山班在读本科生,专业方向:临床医学。E-mail: fq15888947763@126.com
基金资助:
Fangqi BAO1(), Haiye TU2, Mingsun FANG3, Qian ZHANG3, Minli CHEN3()()
Received:2023-09-01
Revised:2023-11-24
Published:2024-04-25
Online:2024-04-25
Contact:
CHEN Minli (ORCID: 0000-0003-1911-9397), E-mail: cmli991@126.com摘要:
尿酸是人类嘌呤代谢的终产物,其过度积累会导致高尿酸血症。高尿酸血症与慢性肾病的关系密切,被认为是后者的独立危险因素,因此由高尿酸血症诱导的慢性肾病也被称为高尿酸肾病。21世纪以来,随着尿酸致病作用研究逐渐深入,以及高尿酸血症动物模型构建的发展,尿酸的致病机制逐渐被揭开,对其诱导慢性肾病的病理生理机制研究也有了重要进展,但对其病理分子机制的认识仍有很大不足。因此,新型的动物模型或造模方式或许能够给高尿酸血症及相关慢性肾病的机制研究提供更好的契机。本文从氧化应激、炎症、自噬、纤维化和肠道微生物等方面介绍高尿酸肾病病理分子机制的研究进展:氧化应激方面,尿酸在细胞内通过黄嘌呤氧化酶、烟酰胺腺嘌呤二核苷酸磷酸氧化酶、线粒体诱导氧化应激并损伤细胞;炎症方面,尿酸晶体可以激活NLRP3炎症小体并启动炎症瀑布,但关于游离尿酸的促炎作用尚存争议;自噬方面,有研究支持促进自噬可缓解尿酸诱导的炎症,也有研究支持完全相反的结论;纤维化方面,上皮间质转化是尿酸引起肾小球硬化和肾小管间质纤维化的重要机制,大量研究寻找了尿酸引起肾组织上皮间质转化的不同信号通路;肠道微生物方面,有益的菌群可通过合成短链脂肪酸、减少尿素肠肝循环、减少尿酸生成保护肾脏。本文旨在帮助人们理解高尿酸血症与慢性肾病之间的复杂关系,为进一步进行相关研究和新药研发提供参考。
中图分类号:
包方奇,屠海烨,方明笋,等. 基于动物模型的高尿酸肾病病理及分子机制研究进展[J]. 实验动物与比较医学, 2024, 44(2): 180-191. DOI: 10.12300/j.issn.1674-5817.2023.123.
Fangqi BAO,Haiye TU,Mingsun FANG,et al. Advances in Research on Pathological and Molecular Mechanism of Hyperuricemic Nephropathy Based on Animal Models[J]. Laboratory Animal and Comparative Medicine, 2024, 44(2): 180-191. DOI: 10.12300/j.issn.1674-5817.2023.123.
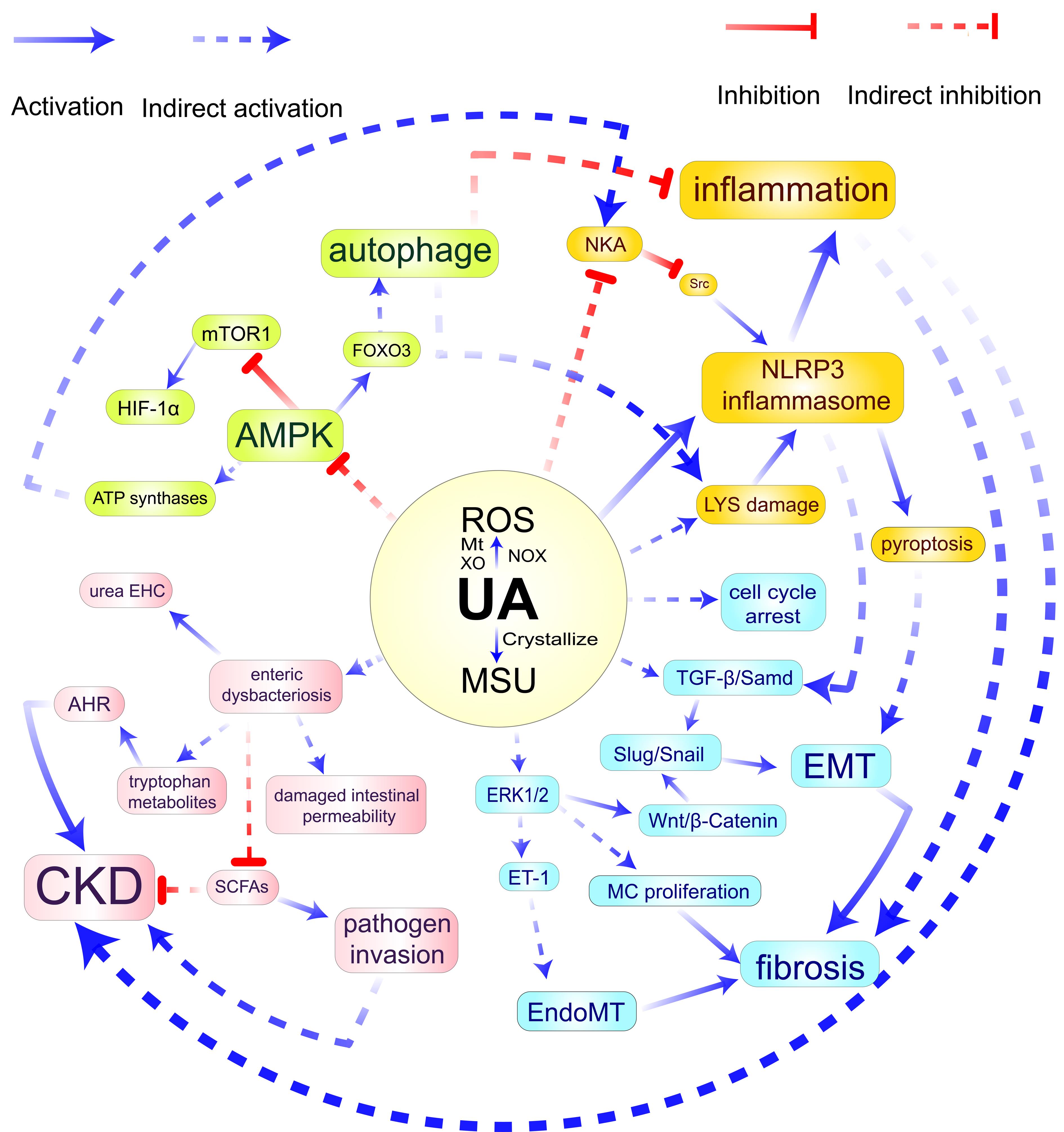
图1 高尿酸肾病的病理学分子机制示意图注:HUA,高尿酸血症;NOX,烟酰胺腺嘌呤二核苷酸磷酸氧化酶;Mt,线粒体;XO,黄嘌呤氧化酶;MSU,单钠尿酸盐;LYS,溶酶体;EMT,上皮间质转化;MC,肾小球系膜细胞;EndoMT,血管内皮细胞间质化;SCFAs,短链脂肪酸;AHR,芳香族氨基酸受体;ET-1,内皮素-1;NKA,钠钾ATP酶;urea EHC,尿素肠肝循环;ROS,活性氧;CKD,慢性肾病。
Figure 1 Pathological molecular mechanism of hyperuricemic nephropathyNote:HUA, Hyperuricemia; NOX, Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase; Mt, Mitochondria; XO, Xanthine oxidase; MSU, Monosodium urate; LYS, Lysosome; EMT, Epithelial-mesenchymal transition; MC, Mesangial cells; EndoMT, Endothelial-to-mesenchymal transition; SCFAs, Short-chain fatty acids; AHR, Aryl Hydrocarbon receptor; ET-1, Endothelin-1; NKA, Sodium-potassium ATPase; urea EHC, Urea enterohepatic circulation; ROS, Reactive oxygen species; CKD, Chronic kidney disease.
| 1 | RIDI R EL, TALLIMA H. Physiological functions and pathogenic potential of uric acid: a review[J]. J Adv Res, 2017, 8(5):487-493. DOI: 10.1016/j.jare.2017.03.003 . |
| 2 | DEMIRAY A, AFSAR B, COVIC A, et al. The role of uric acid in the acute myocardial infarction: a narrative review[J]. Angiology, 2022, 73(1):9-17. DOI: 10.1177/00033197211012546 . |
| 3 | ZOPPINI G, TARGHER G, CHONCHOL M, et al. Serum uric acid levels and incident chronic kidney disease in patients with type 2 diabetes and preserved kidney function[J]. Diabetes Care, 2012, 35(1):99-104. DOI: 10.2337/dc11-1346 . |
| 4 | BADVE S V, PASCOE E M, TIKU A, et al. Effects of allopurinol on the progression of chronic kidney disease[J]. N Engl J Med, 2020, 382(26):2504-2513. DOI: 10.1056/NEJMoa1915833 . |
| 5 | DORIA A, GALECKI A T, SPINO C, et al. Serum urate lowering with allopurinol and kidney function in type 1 diabetes[J]. N Engl J Med, 2020, 382(26):2493-2503. DOI: 10.1056/NEJMoa1916624 . |
| 6 | BELLOMO G, VENANZI S, VERDURA C, et al. Association of uric acid with change in kidney function in healthy normotensive individuals[J]. Am J Kidney Dis, 2010, 56(2):264-272. DOI: 10.1053/j.ajkd.2010.01.019 . |
| 7 | GOLDBERG A, GARCIA-ARROYO F, SASAI F, et al. Mini review: reappraisal of uric acid in chronic kidney disease[J]. Am J Nephrol, 2021, 52(10-11):837-844. DOI: 10.1159/000519491 . |
| 8 | SATO Y, FEIG D I, STACK A G, et al. The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD[J]. Nat Rev Nephrol, 2019, 15(12):767-775. DOI: 10.1038/s41581-019-0174-z . |
| 9 | LU J, DALBETH N, YIN H Y, et al. Mouse models for human hyperuricaemia: a critical review[J]. Nat Rev Rheumatol, 2019, 15(7):413-426. DOI: 10.1038/s41584-019-0222-x . |
| 10 | YANAI H, ADACHI H, HAKOSHIMA M, et al. Molecular biological and clinical understanding of the pathophysiology and treatments of hyperuricemia and its association with metabolic syndrome, cardiovascular diseases and chronic kidney disease[J]. Int J Mol Sci, 2021, 22(17):9221. DOI: 10.3390/ijms22179221 . |
| 11 | MACÍAS N, GOICOECHEA M, DE VINUESA M S, et al. Urate reduction and renal preservation: what is the evidence?[J]. Curr Rheumatol Rep, 2013, 15(12):386. DOI: 10.1007/s11926-013-0386-3 . |
| 12 | RAFEY M A, LIPKOWITZ M S, LEAL-PINTO E, et al. Uric acid transport[J]. Curr Opin Nephrol Hypertens, 2003, 12(5):511-516. DOI: 10.1097/00041552-200309000-00005 . |
| 13 | EJAZ A A, NAKAGAWA T, KANBAY M, et al. Hyperuricemia in kidney disease: a major risk factor for cardiovascular events, vascular calcification, and renal damage[J]. Semin Nephrol, 2020, 40(6):574-585. DOI: 10.1016/j.semnephrol.2020.12.004 . |
| 14 | MCCORMICK N, O'CONNOR M J, YOKOSE C, et al. Assessing the causal relationships between insulin resistance and hyperuricemia and gout using bidirectional Mendelian randomization[J]. Arthritis Rheumatol, 2021, 73(11):2096-2104. DOI: 10.1002/art.41779 . |
| 15 | RAMOS G K, GOLDFARB D S. Update on uric acid and the kidney[J]. Curr Rheumatol Rep, 2022, 24(5):132-138. DOI: 10.1007/s11926-022-01069-3 . |
| 16 | 张璐, 杨定位. 高尿酸血症肾病的诊治进展[J]. 中华临床医师杂志(电子版), 2019, 13(6):457-462. DOI: 10.3877/cma.j.issn.1674-0785.2019.06.011 . |
| ZHANG L, YANG D W. Progress in diagnosis and treatment of hyperuricemic nephropathy[J]. Chin J Clin Electron Ed, 2019, 13(6):457-462. DOI: 10.3877/cma.j.issn.1674-0785.2019.06.011 . | |
| 17 | 卢忠英, 郁建平, 朱梦琪, 等. 不同组合造模剂诱导大鼠高尿酸血症模型的比较研究[J]. 山地农业生物学报, 2014, 33(5):40-42, 67. DOI: 10.15958/j.cnki.sdnyswxb.2014.05.009 . |
| LU Z Y, YU J P, ZHU M Q, et al. Comparative study of different combinations of modeling agent-induced rat model of hyperuricemia[J]. J Mt Agric Biol, 2014, 33(5):40-42, 67. DOI: 10.15958/j.cnki.sdnyswxb.2014.05.009 . | |
| 18 | DEBOSCH B J, KLUTH O, FUJIWARA H, et al. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9[J]. Nat Commun, 2014, 5:4642. DOI: 10.1038/ncomms5642 . |
| 19 | ZHU L R, DONG Y F, NA S, et al. Saponins extracted from Dioscorea collettii rhizomes regulate the expression of urate transporters in chronic hyperuricemia rats[J]. Biomed Pharmacother, 2017, 93:88-94. DOI: 10.1016/j.biopha.2017.06.022 . |
| 20 | SALEM C BEN, SLIM R, FATHALLAH N, et al. Drug-induced hyperuricaemia and gout[J]. Rheumatology, 2017, 56(5):679-688. DOI: 10.1093/rheumatology/kew293 . |
| 21 | HONG F, ZHENG A J, XU P F, et al. High-protein diet induces hyperuricemia in a new animal model for studying human gout[J]. Int J Mol Sci, 2020, 21(6):2147. DOI: 10.3390/ijms21062147 . |
| 22 | SAUTIN Y Y, NAKAGAWA T, ZHARIKOV S, et al. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress[J]. Am J Physiol Cell Physiol, 2007, 293(2): C584-C596. DOI: 10.1152/ajpcell.00600.2006 . |
| 23 | KIMURA Y, TSUKUI D, KONO H. Uric acid in inflammation and the pathogenesis of atherosclerosis[J]. Int J Mol Sci, 2021, 22(22):12394. DOI: 10.3390/ijms222212394 . |
| 24 | WAHEED Y, YANG F, SUN D. Role of asymptomatic hyperuricemia in the progression of chronic kidney disease and cardiovascular disease[J]. Korean J Intern Med, 2021, 36(6):1281-1293. DOI: 10.3904/kjim.2020.340 . |
| 25 | GHERGHINA M E, PERIDE I, TIGLIS M, et al. Uric acid and oxidative stress-relationship with cardiovascular, metabolic, and renal impairment[J]. Int J Mol Sci, 2022, 23(6):3188. DOI: 10.3390/ijms23063188 . |
| 26 | ELEFTHERIADIS T, PISSAS G, ANTONIADI G, et al. Allopurinol protects human glomerular endothelial cells from high glucose-induced reactive oxygen species generation, p53 overexpression and endothelial dysfunction[J]. Int Urol Nephrol, 2018, 50(1):179-186. DOI: 10.1007/s11255-017-1733-5 . |
| 27 | CHAO H H, LIU J C, LIN J W, et al. Uric acid stimulates endothelin-1 gene expression associated with NADPH oxidase in human aortic smooth muscle cells[J]. Acta Pharmacol Sin, 2008, 29(11):1301-1312. DOI: 10.1111/j.1745-7254.2008.00877.x . |
| 28 | ZHUANG Y B, FENG Q C, DING G X, et al. Activation of ERK1/2 by NADPH oxidase-originated reactive oxygen species mediates uric acid-induced mesangial cell proliferation[J]. Am J Physiol Renal Physiol, 2014, 307(4): F396-F406. DOI: 10.1152/ajprenal.00565.2013 . |
| 29 | CRISTÓBAL-GARCÍA M, GARCÍA-ARROYO F E, TAPIA E, et al. Renal oxidative stress induced by long-term hyperuricemia alters mitochondrial function and maintains systemic hypertension[J]. Oxid Med Cell Longev, 2015, 2015:535686. DOI: 10.1155/2015/535686 . |
| 30 | HONG Q, QI K, FENG Z, et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload[J]. Cell Calcium, 2012, 51(5):402-410. DOI: 10.1016/j.ceca.2012.01.003 . |
| 31 | SU Y, HU L T, WANG Y N, et al. The Rho kinase signaling pathway participates in tubular mitochondrial oxidative injury and apoptosis in uric acid nephropathy[J]. J Int Med Res, 2021, 49(6):3000605211021752. DOI: 10.1177/03000605211021752 . |
| 32 | YANG L J, CHANG B C, GUO Y L, et al. The role of oxidative stress-mediated apoptosis in the pathogenesis of uric acid nephropathy[J]. Ren Fail, 2019, 41(1):616-622. DOI: 10.1080/0886022X.2019.1633350 . |
| 33 | BRAGA T T, FORNI M F, CORREA-COSTA M, et al. Soluble uric acid activates the NLRP3 inflammasome[J]. Sci Rep, 2017, 7:39884. DOI: 10.1038/srep39884 . |
| 34 | KOMORI H, YAMADA K, TAMAI I. Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells[J]. Biochim Biophys Acta Biomembr, 2018, 1860(5):973-980. DOI: 10.1016/j.bbamem.2018.01.006 . |
| 35 | BROVOLD H, LUND T, SVISTOUNOV D, et al. Crystallized but not soluble uric acid elicits pro-inflammatory response in short-term whole blood cultures from healthy men[J]. Sci Rep, 2019, 9:10513. DOI: 10.1038/s41598-019-46935-w . |
| 36 | SU H Y, YANG C, LIANG D, et al. Research advances in the mechanisms of hyperuricemia-induced renal injury[J]. Biomed Res Int, 2020, 2020:5817348. DOI: 10.1155/2020/5817348 . |
| 37 | YE Y, ZHANG Y, WANG B, et al. CXCR1/CXCR2 antagonist G31P inhibits nephritis in a mouse model of uric acid nephropathy[J]. Biomed Pharmacother, 2018, 107:1142-1150. DOI: 10.1016/j.biopha.2018.07.077 . |
| 38 | CHOE J Y, CHOI C H, PARK K Y, et al. High-mobility group box 1 is responsible for monosodium urate crystal-induced inflammation in human U937 macrophages[J]. Biochem Biophys Res Commun, 2018, 503(4):3248-3255. DOI: 10.1016/j.bbrc.2018.08.139 . |
| 39 | VÄLIMÄKI E, MIETTINEN J J, LIETZÉN N, et al. Monosodium urate activates Src/Pyk2/PI3 kinase and cathepsin dependent unconventional protein secretion from human primary macrophages[J]. Mol Cell Proteomics, 2013, 12(3):749-763. DOI: 10.1074/mcp.M112.024661 . |
| 40 | WANG M, LIN X, YANG X M, et al. Research progress on related mechanisms of uric acid activating NLRP3 inflammasome in chronic kidney disease[J]. Ren Fail, 2022, 44(1):615-624. DOI: 10.1080/0886022X.2022.2036620 . |
| 41 | ISAKA Y, TAKABATAKE Y, TAKAHASHI A, et al. Hyperuricemia-induced inflammasome and kidney diseases[J]. Nephrol Dial Transplant, 2016, 31(6):890-896. DOI: 10.1093/ndt/gfv024 . |
| 42 | WANG G H, ZUO T, LI R. The mechanism of Arhalofenate in alleviating hyperuricemia-activating PPARγ thereby reducing caspase-1 activity[J]. Drug Dev Res, 2020, 81(7):859-866. DOI: 10.1002/ddr.21699 . |
| 43 | ELEFTHERIADIS T, PISSAS G, SOUNIDAKI M, et al. Urate crystals directly activate the T-cell receptor complex and induce T-cell proliferation[J]. Biomed Rep, 2017, 7(4):365-369. DOI: 10.3892/br.2017.960 . |
| 44 | KOKA R M, HUANG E, LIESKE J C. Adhesion of uric acid crystals to the surface of renal epithelial cells[J]. Am J Physiol Renal Physiol, 2000, 278(6): F989-F998. DOI: 10.1152/ajprenal.2000.278.6.F989 . |
| 45 | JOOSTEN L A B, CRIŞAN T O, BJORNSTAD P, et al. Asymptomatic hyperuricaemia: a silent activator of the innate immune system[J]. Nat Rev Rheumatol, 2020, 16(2):75-86. DOI: 10.1038/s41584-019-0334-3 . |
| 46 | ZHOU R B, TARDIVEL A, THORENS B, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation[J]. Nat Immunol, 2010, 11(2):136-140. DOI: 10.1038/ni.1831 . |
| 47 | MISAWA T, TAKAHAMA M, KOZAKI T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome[J]. Nat Immunol, 2013, 14(5):454-460. DOI: 10.1038/ni.2550 . |
| 48 | ELLIOTT E I, MILLER A N, BANOTH B, et al. Cutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming[J]. J Immunol, 2018, 200(9):3047-3052. DOI: 10.4049/jimmunol.1701723 . |
| 49 | XIAO J, ZHANG X L, FU C S, et al. Soluble uric acid increases NALP3 inflammasome and interleukin-1β expression in human primary renal proximal tubule epithelial cells through the Toll-like receptor 4-mediated pathway[J]. Int J Mol Med, 2015, 35(5):1347-1354. DOI: 10.3892/ijmm.2015.2148 . |
| 50 | CRIȘAN T O, CLEOPHAS M C, OOSTING M, et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra[J]. Ann Rheum Dis, 2016, 75(4):755-762. DOI: 10.1136/annrheumdis-2014-206564 . |
| 51 | KIMURA Y, YANAGIDA T, ONDA A, et al. Soluble uric acid promotes atherosclerosis via AMPK (AMP-activated protein kinase)-mediated inflammation[J]. Arterioscler Thromb Vasc Biol, 2020, 40(3):570-582. DOI: 10.1161/ATVBAHA.119.313224 . |
| 52 | DE FRANCESCO E M, MAGGIOLINI M, MUSTI A M. Crosstalk between Notch, HIF-1α and GPER in breast cancer EMT[J]. Int J Mol Sci, 2018, 19(7):2011. DOI: 10.3390/ijms19072011 . |
| 53 | YANG Q M, FU C S, ZHANG X L, et al. Adiponectin protects against uric acid-induced renal tubular epithelial inflammatory responses via the AdipoR1/AMPK signaling pathway[J]. Int J Mol Med, 2019, 43(3):1542-1552. DOI: 10.3892/ijmm.2019.4072 . |
| 54 | YIN W, ZHOU Q L, OUYANG S X, et al. Uric acid regulates NLRP3/IL-1β signaling pathway and further induces vascular endothelial cells injury in early CKD through ROS activation and K+ efflux[J]. BMC Nephrol, 2019, 20(1):319. DOI: 10.1186/s12882-019-1506-8 . |
| 55 | XIAO J, ZHANG X L, FU C S, et al. Impaired Na+-K+-ATPase signaling in renal proximal tubule contributes to hyperuricemia-induced renal tubular injury[J]. Exp Mol Med, 2018, 50(3): e452. DOI: 10.1038/emm.2017.287 . |
| 56 | XIAO J, ZHU S B, GUAN H C, et al. AMPK alleviates high uric acid-induced Na+-K+-ATPase signaling impairment and cell injury in renal tubules[J]. Exp Mol Med, 2019, 51(5):1-14. DOI: 10.1038/s12276-019-0254-y . |
| 57 | MA Q Y, IMMLER R, PRUENSTER M, et al. Soluble uric acid inhibits β2 integrin-mediated neutrophil recruitment in innate immunity[J]. Blood, 2022, 139(23):3402-3417. DOI: 10.1182/blood.2021011234 . |
| 58 | SELLMAYR M, HERNANDEZ PETZSCHE M R, MA Q Y, et al. Only hyperuricemia with crystalluria, but not asymptomatic hyperuricemia, drives progression of chronic kidney disease[J]. J Am Soc Nephrol, 2020, 31(12):2773-2792. DOI: 10.1681/ASN.2020040523 . |
| 59 | ALBERTS B M, BARBER J S, SACRE S M, et al. Precipitation of soluble uric acid is necessary for in vitro activation of the NLRP3 inflammasome in primary human monocytes[J]. J Rheumatol, 2019, 46(9):1141-1150. DOI: 10.3899/jrheum.180855 . |
| 60 | MA Q Y, HONARPISHEH M, LI C Y, et al. Soluble uric acid is an intrinsic negative regulator of monocyte activation in monosodium urate crystal-induced tissue inflammation[J]. J Immunol, 2020, 205(3):789-800. DOI: 10.4049/jimmunol. 2000319 . |
| 61 | CHOI A M K, RYTER S W, LEVINE B. Autophagy in human health and disease[J]. N Engl J Med, 2013, 368(7):651-662. DOI: 10.1056/NEJMra1205406 . |
| 62 | SHI C S, SHENDEROV K, HUANG N N, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction[J]. Nat Immunol, 2012, 13(3):255-263. DOI: 10.1038/ni.2215 . |
| 63 | BAO J F, SHI Y F, TAO M, et al. Pharmacological inhibition of autophagy by 3-MA attenuates hyperuricemic nephropathy[J]. Clin Sci, 2018, 132(21):2299-2322. DOI: 10.1042/CS20180563 . |
| 64 | HU J C, WU H, WANG D C, et al. Weicao capsule ameliorates renal injury through increasing autophagy and NLRP3 degradation in UAN rats[J]. Int J Biochem Cell Biol, 2018, 96:1-8. DOI: 10.1016/j.biocel.2018.01.001 . |
| 65 | HERZIG S, SHAW R J. AMPK: guardian of metabolism and mitochondrial homeostasis[J]. Nat Rev Mol Cell Biol, 2018, 19(2):121-135. DOI: 10.1038/nrm.2017.95 . |
| 66 | HU Y, SHI Y F, CHEN H, et al. Blockade of autophagy prevents the progression of hyperuricemic nephropathy through inhibiting NLRP3 inflammasome-mediated pyroptosis[J]. Front Immunol, 2022, 13:858494. DOI: 10.3389/fimmu. 2022. 858494 . |
| 67 | MAEJIMA I, TAKAHASHI A, OMORI H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury[J]. EMBO J, 2013, 32(17):2336-2347. DOI: 10.1038/emboj.2013.171 . |
| 68 | COELHO S C, BERILLO O, CAILLON A, et al. Three-month endothelial human endothelin-1 overexpression causes blood pressure elevation and vascular and kidney injury[J]. Hypertension, 2018, 71(1):208-216. DOI: 10.1161/HYPERTENSIONAHA.117.09925 . |
| 69 | YANG X L, GU J, LV H C, et al. Uric acid induced inflammatory responses in endothelial cells via up-regulating(pro)renin receptor[J]. Biomedecine Pharmacother, 2019, 109:1163-1170. DOI: 10.1016/j.biopha.2018.10.129 . |
| 70 | REYES-MARTINEZ C, NGUYEN Q M, KASSAN M, et al. (pro)renin receptor-dependent induction of profibrotic factors is mediated by COX-2/EP4/NOX-4/smad pathway in collecting duct cells[J]. Front Pharmacol, 2019, 10:803. DOI: 10.3389/fphar.2019.00803 . |
| 71 | KO J, KANG H J, KIM D A, et al. Uric acid induced the phenotype transition of vascular endothelial cells via induction of oxidative stress and glycocalyx shedding[J]. FASEB J, 2019, 33(12):13334-13345. DOI: 10.1096/fj.201901148R . |
| 72 | ALBERTONI G, MAQUIGUSSA E, PESSOA E, et al. Soluble uric acid increases intracellular calcium through an angiotensin II-dependent mechanism in immortalized human mesangial cells[J]. Exp Biol Med, 2010, 235(7):825-832. DOI: 10.1258/ebm.2010.010007 . |
| 73 | RYU E S, KIM M J, SHIN H S, et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease[J]. Am J Physiol Renal Physiol, 2013, 304(5): F471-F480. DOI: 10.1152/ajprenal. 00560. 2012 . |
| 74 | LOVISA S, ZEISBERG M, KALLURI R. Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis[J]. Trends Endocrinol Metab, 2016, 27(10):681-695. DOI: 10.1016/j.tem.2016.06.004 . |
| 75 | REN Q, TAO S B, GUO F, et al. Natural flavonol fisetin attenuated hyperuricemic nephropathy via inhibiting IL-6/JAK2/STAT3 and TGF-β/SMAD3 signaling[J]. Phytomedicine, 2021, 87:153552. DOI: 10.1016/j.phymed.2021.153552 . |
| 76 | WANG W J, WANG X Y, CHUN J, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium[J]. J Immunol, 2013, 190(3):1239-1249. DOI: 10.4049/jimmunol.1201959 . |
| 77 | ROMERO C A, REMOR A, LATINI A, et al. Uric acid activates NRLP3 inflammasome in an in-vivo model of epithelial to mesenchymal transition in the kidney[J]. J Mol Histol, 2017, 48(3):209-218. DOI: 10.1007/s10735-017-9720-9 . |
| 78 | TAO M, SHI Y F, TANG L X, et al. Blockade of ERK1/2 by U0126 alleviates uric acid-induced EMT and tubular cell injury in rats with hyperuricemic nephropathy[J]. Am J Physiol Renal Physiol, 2019, 316(4): F660-F673. DOI: 10.1152/ajprenal. 00480.2018 . |
| 79 | ZHA D Q, WU S Q, GAO P, et al. Telmisartan attenuates uric acid-induced epithelial-mesenchymal transition in renal tubular cells[J]. Biomed Res Int, 2019, 2019:3851718. DOI: 10.1155/2019/3851718 . |
| 80 | SCHUNK S J, FLOEGE J, FLISER D, et al. WNT-β-catenin signalling―a versatile player in kidney injury and repair[J]. Nat Rev Nephrol, 2021, 17(3):172-184. DOI: 10.1038/s41581-020-00343-w . |
| 81 | ZHAO L, LI C Y, ZHOU B, et al. Crucial role of serum response factor in renal tubular epithelial cell epithelial-mesenchymal transition in hyperuricemic nephropathy[J]. Aging, 2019, 11(22):10597-10609. DOI: 10.18632/aging.102479 . |
| 82 | SHI Y F, XU L Q, TAO M, et al. Blockade of enhancer of zeste homolog 2 alleviates renal injury associated with hyperuricemia[J]. Am J Physiol Renal Physiol, 2019, 316(3): F488-F505. DOI: 10.1152/ajprenal.00234.2018 . |
| 83 | ALMEIDA A, MITCHELL A L, BOLAND M, et al. A new genomic blueprint of the human gut microbiota[J]. Nature, 2019, 568(7753):499-504. DOI: 10.1038/s41586-019-0965-1 . |
| 84 | STAVROPOULOU E, KANTARTZI K, TSIGALOU C, et al. Focus on the gut-kidney axis in health and disease[J]. Front Med, 2021, 7:620102. DOI: 10.3389/fmed.2020.620102 . |
| 85 | WEI J, ZHANG Y Q, DALBETH N, et al. Association between gut microbiota and elevated serum urate in two independent cohorts[J]. Arthritis Rheumatol, 2022, 74(4):682-691. DOI: 10.1002/art.42009 . |
| 86 | WANG J, CHEN Y, ZHONG H, et al. The gut microbiota as a target to control hyperuricemia pathogenesis: potential mechanisms and therapeutic strategies[J]. Crit Rev Food Sci Nutr, 2022, 62(14):3979-3989. DOI: 10.1080/10408398. 2021. 1874287 . |
| 87 | NIEUWDORP M, GILIJAMSE P W, PAI N, et al. Role of the microbiome in energy regulation and metabolism[J]. Gastroenterology, 2014, 146(6):1525-1533. DOI: 10.1053/j.gastro.2014.02.008 . |
| 88 | HE Y, FU L H, LI Y P, et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8+ T cell immunity[J]. Cell Metab, 2021, 33(5):988-1000.e7. DOI: 10.1016/j.cmet.2021.03.002 . |
| 89 | PAN L B, HAN P, MA S R, et al. Abnormal metabolism of gut microbiota reveals the possible molecular mechanism of nephropathy induced by hyperuricemia[J]. Acta Pharm Sin B, 2020, 10(2):249-261. DOI: 10.1016/j.apsb.2019.10.007 . |
| 90 | XU D X, LV Q L, WANG X F, et al. Hyperuricemia is associated with impaired intestinal permeability in mice[J]. Am J Physiol Gastrointest Liver Physiol, 2019, 317(4): G484-G492. DOI: 10.1152/ajpgi.00151.2019 . |
| 91 | ZHAO H Y, CHEN X Y, ZHANG L, et al. Lacticaseibacillus rhamnosus Fmb14 prevents purine induced hyperuricemia and alleviate renal fibrosis through gut-kidney axis[J]. Pharmacol Res, 2022, 182:106350. DOI: 10.1016/j.phrs. 2022.106350 . |
| 92 | LIU X, LV Q L, REN H Y, et al. The altered gut microbiota of high-purine-induced hyperuricemia rats and its correlation with hyperuricemia[J]. PeerJ, 2020, 8: e8664. DOI: 10.7717/peerj.8664 . |
| 93 | NUKI G. An appraisal of the 2012 American college of rheumatology guidelines for the management of gout[J]. Curr Opin Rheumatol, 2014, 26(2):152-161. DOI: 10.1097/BOR. 0000000000000034 . |
| 94 | WHITE W B, SAAG K G, BECKER M A, et al. Cardiovascular safety of febuxostat or allopurinol in patients with gout[J]. N Engl J Med, 2018, 378(13):1200-1210. DOI: 10.1056/NEJMoa1710895 . |
| 95 | LIU P, WANG C, WANG Y, et al. Zishen Qingre Tongluo formula improves renal fatty acid oxidation and alleviated fibrosis via the regulation of the TGF- β1/Smad3 signaling pathway in hyperuricemic nephrology rats[J]. Biomed Res Int, 2021, 2021:2793823. DOI: 10.1155/2021/2793823 . |
| 96 | LI X Q, CHEN Y H, GAO X X, et al. Antihyperuricemic effect of green Alga Ulva lactuca ulvan through regulating urate transporters[J]. J Agric Food Chem, 2021, 69(38):11225-11235. DOI: 10.1021/acs.jafc.1c03607 . |
| 97 | WU D, CHEN R H, LI Q H, et al. Tea (Camellia sinensis) ameliorates hyperuricemia via uric acid metabolic pathways and gut microbiota[J]. Nutrients, 2022, 14(13):2666. DOI: 10.3390/nu14132666 . |
| 98 | WEN X H, LOU Y, SONG S Y, et al. Qu-Zhuo-Tong-Bi Decoction alleviates gouty arthritis by regulating butyrate-producing bacteria in mice[J]. Front Pharmacol, 2021, 11:610556. DOI: 10.3389/fphar.2020.610556 . |
| [1] | 刘亚益, 贾云凤, 左一鸣, 张军平, 吕仕超. 心气阴两虚证动物模型的构建方法与评价进展[J]. 实验动物与比较医学, 2025, 45(4): 411-421. |
| [2] | 赵鑫, 王晨曦, 石文清, 娄月芬. 斑马鱼在炎症性肠病机制及药物研究中的应用进展[J]. 实验动物与比较医学, 2025, 45(4): 422-431. |
| [3] | 李会萍, 高洪彬, 温金银, 杨锦淳. 疾病动物模型数字化图谱数据库平台的构建与初步应用[J]. 实验动物与比较医学, 2025, 45(3): 300-308. |
| [4] | 潘颐聪, 蒋汶洪, 胡明, 覃晓. 慢性肾脏病大鼠主动脉钙化模型的术式优化及效果评价[J]. 实验动物与比较医学, 2025, 45(3): 279-289. |
| [5] | 陈钰涵, 陈瑾玲, 李欣, 区燕华, 王斯, 陈镜伊, 王兴易, 袁嘉丽, 段媛媛, 羊忠山, 牛海涛. 基于中西医临床病证特点的重症肌无力动物模型分析[J]. 实验动物与比较医学, 2025, 45(2): 176-186. |
| [6] | 连辉, 姜艳玲, 刘佳, 张玉立, 谢伟, 薛晓鸥, 李健. 异常子宫出血大鼠模型的构建与评价[J]. 实验动物与比较医学, 2025, 45(2): 130-146. |
| [7] | 罗世雄, 张赛, 陈慧. 常见哮喘动物模型的建立方法与评价研究进展[J]. 实验动物与比较医学, 2025, 45(2): 167-175. |
| [8] | 王碧莹, 鲁家铄, 昝桂影, 陈若松, 柴景蕊, 刘景根, 王瑜珺. 啮齿类动物药物成瘾模型的构建方法和应用进展[J]. 实验动物与比较医学, 2025, 45(2): 158-166. |
| [9] | 费彬, 郭文科, 郭建平. 疝疾病动物模型研究及新型疝修补材料应用进展[J]. 实验动物与比较医学, 2025, 45(1): 55-66. |
| [10] | 杨家豪, 丁纯蕾, 钱风华, 孙旗, 姜旭升, 陈雯, 沈梦雯. 脓毒症相关脏器损伤动物模型研究进展[J]. 实验动物与比较医学, 2024, 44(6): 636-644. |
| [11] | 孙效容, 苏丹, 贵文娟, 陈玥. 手术诱导大鼠中重度膝骨关节炎模型的建立与评价[J]. 实验动物与比较医学, 2024, 44(6): 597-604. |
| [12] | 田芳, 潘滨, 史佳怡, 徐燕意, 李卫华. 大气细颗粒物PM2.5暴露动物模型建立方法及在生殖毒性研究中的应用进展[J]. 实验动物与比较医学, 2024, 44(6): 626-635. |
| [13] | 赵小娜, 王鹏, 叶茂青, 曲新凯. 应用Triacsin C构建新型高血糖肥胖小鼠心功能减退模型[J]. 实验动物与比较医学, 2024, 44(6): 605-612. |
| [14] | 涂颖欣, 纪依澜, 王菲, 杨东明, 王冬冬, 孙芷馨, 戴悦欣, 王言吉, 阚广捍, 吴斌, 赵德明, 杨利峰. 小型猪后肢去负荷模拟失重模型的建立与组织损伤研究[J]. 实验动物与比较医学, 2024, 44(5): 475-486. |
| [15] | 黄冬妍, 吴建辉. 生殖毒理学研究动物模型的建立方法及应用评价[J]. 实验动物与比较医学, 2024, 44(5): 550-559. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||